

{kind=link}
Animals and treatment
A total of 35 healthy female specific pathogen-free (SPF) Wistar rats aged 6–8 weeks and weighing 200 ± 20 g were purchased from Vital River Laboratories (Beijing, China). All animal experiments were reviewed and approved by the Ethics Committee of Sichuan University West China Medical for Animal Experimentation (K2019002-2). All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and complied with the requirements of the National Act on the Use of Experimental Animals (China). The authors followed the ARRIVE guidelines to minimize animal suffering.
After 1 week of adaptive feeding, 10 rats were selected for the blank group according to the random number table method, and the remaining 25 rats were intradermally immunized with 200 μg of cattle type II collagen (Chondrex, Redmond, WA, USA) emulsified in incomplete Freund’s adjuvant (Chondrex) at the base of the tail and then given a booster immunization 7 days later to establish the CIA model. The blank group of rats were subcutaneously injected with the same volume of sterile saline.
Finally, the model was successfully established in 22 rats, and these rats were randomly divided into the control group and the ATRA group. We utilized the following three groups: the blank group (healthy Wistar rats gavaged with corn oil three times a week), the control group (CIA model Wistar rats gavaged with corn oil three times a week) and the ATRA group(CIA model Wistar rats gavaged with 1 mg/kg ATRA (Sigma‒Aldrich, St. Louis, MO, USA) dissolved in corn oil three times a week). The body weight and the arthritis index (AI)28,29 were measured and recorded every week. The thickness of the paw was measured using callipers 9 weeks after the primary immunization. The rats were sacrificed with anaesthesia using ketamine/xylazine (100/10 mg/kg) after 6 weeks of treatment. The entire process is shown in Fig. 1.
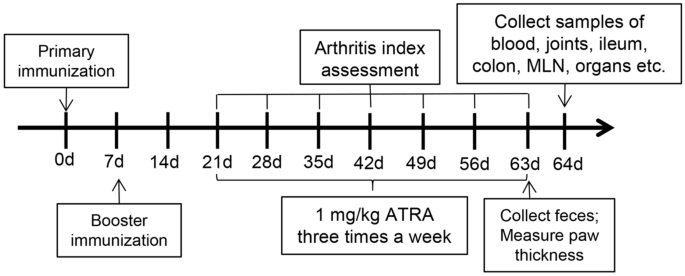
Experimental study design.
Histopathology and immunohistochemistry (IHC)
Ankle joints, livers, kidneys, ileums, colons, etc., were fixed with formalin and embedded in paraffin, and ankle joints were further decalcified with 10% ethylenediaminetetraacetic acid (EDTA) solution for 8 weeks. Then, the paraffin-embedded specimens were cut into serial sections (4 μm thick) and stained with haematoxylin and eosin (H&E). Images of three fields of view were randomly captured using a microscope (Leica, Wetzlar, Germany).
Slides of ankle joints, ileums and colons were scored blindly by 2 individuals. The following scoring system was used to assess the ankle joints: 0 = normal joints, absence of inflammation; 1 = evidence of soft tissue inflammation, synovial hyperplasia and/or cell infiltration into the synovial space (pannus); 2 = Grade 1 and definite erosion of articular cartilage; 3 = Grade 2 and definite erosion of articular bone, but joint architecture mostly intact; and 4 = Grade 3 with bone erosion resulting in a major loss of joint architecture30. The histological score for ileums and colons were determined based on a scale that graded the extent of inflammatory infiltrate (0–5), crypt damage (0–4), ulceration (0–3), and the presence or absence of edema (0 or 1)31.
Sections were then deparaffinized, rehydrated through graded ethanol solutions, and subjected to endogenous peroxidase activity blocking. Claudin (1:250 dilution, Bioss, Beijing, China), Occludin (1:600 dilution, Servicebio, Wuhan, Hubei, China), and ZO-1 primary antibodies (1:250 dilution, Bioss, Beijing, China) were applied in a humidified chamber at room temperature overnight. Afterwards, the sections were incubated with secondary antibody (1:500 dilution, Biosharp, Hefei, Anhui, China) at room temperature for 30 min. Subsequently, sections were processed with SABC, incubated at room temperature for 30 min, treated with DAB colour solution, and rinsed with water. Haematoxylin staining was performed for 2 min, followed by dehydration in graded alcohol solutions, and slides were sealed with neutral gum. Immunostained sections were examined and imaged under a light microscope at 200 × magnification, and ImageJ software (http://rsb.Info.nih.gov/ij) was applied for quantitative measurement by randomly selecting three fields in every slide. The mean optical density value was calculated by dividing the cumulative optical density by the total area.
Western blotting analysis
To extract cellular and tissue proteins, RIPA buffer supplemented with protease inhibitor cocktail (Beyotime, Shanghai, China) was used to lyse samples at 4 °C for 30 min. Equal amounts of protein were loaded onto SDS‒PAGE gels and then transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking the membranes with 5% non-fat milk at room temperature for 1 h, primary antibodies were added and the samples were incubated overnight at 4 °C; then, the samples were incubation with peroxidase-conjugated secondary antibodies at room temperature for 1 h. Immunoreactive proteins were analysed using an enhanced chemiluminescence kit (Millipore). Quantitation of the relative intensity of western blotting bands was performed using ImageJ software. Western blotting analysis were performed in technical triplicates.
Enzyme-linked immunosorbent assay (ELISA)
Whole blood was collected from the rats and left at room temperature for 2 h. After coagulation, the blood was centrifuged at 3000 rpm for 10 min to obtain serum, which was then stored at − 80 °C until further use. The dissected ileal and colon tissues were washed with saline, dried using filter paper, and weighed. These intestinal tissues were then cut into pieces to create a 10.00% tissue homogenate (intestinal tissue (g): cell lysate (mL) = 1:9) with cell lysate (Boster, Wuhan, China) in an ice bath. After centrifugation at 8000 rpm for 10 min at 4 °C, the supernatant was collected and stored at − 80 °C for future use. The concentrations of tumour necrosis factor-α (TNF-α), interleukin (IL)-6, IL-17, IL-10, secretory immunoglobulin A (SIgA), vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor 2 (VEGFR2) were determined using ELISA kits (4A Biotech, Beijing, China). The absorbance at 450 nm was immediately measured in each well using a Multiskan Spectrum (Bio-Rad, California, USA). Sample concentrations were calculated based on the standard curve. ELISA was performed on each sample in triplicate.
Transmission electron microscopy (TEM) analysis
Five-millimetre segments of fresh ileum and colon tissues were flushed with PBS and fixed in 3% glutaraldehyde at 4 °C for 4 h. After rinsing in PBS, the tissue underwent additional fixation in PBS containing 1% osmium tetroxide (Seebio, Shanghai, China) for 2 h at room temperature, followed by further PBS rinsing and dehydration. Next, the tissues were embedded in Epon 812 (SPI, West Chester, PA, USA) and left to cure in an oven at 60 °C for 48 h. Sections with a thickness of 80 nm were cut using a diamond knife on an ultramicrotome (EM UC7, Leica). These sections were placed on single-hole grids coated with Formvar and carbon and then double-stained in aqueous solutions of 8% uranyl acetate for 25 min at 60 °C and lead citrate for 3 min at room temperature. Images of three fields of view were randomly captured using a JEM-1400 FLASH Transmission Electron Microscope (JEOL, Tokyo, Japan).
Flow cytometry
Whole blood and mesenteric lymph nodes (MLNs) were collected from the rats. MLNs were mechanically mashed and filtered through a 100-mm nylon strainer to prepare a single-cell suspension. Mononuclear cells in blood were extracted using rat lymphocyte separation medium (Hao Yang Biological Technology, Tianjin, China). For the T helper 17 (Th17) assays, cells were stimulated with Cell Stimulation Cocktail (eBioscience, San Diego, CA, USA) for 6 h at 37 °C in a 5% CO2 environment and subsequently stained with CD4-phycoerythrin (PE) antibody. Fixation and permeabilization were performed using fix/perm buffer (Servicebio, Wuhan, China), followed by incubation with an IL-17-allophycocyanin (APC) antibody. For the Treg assay, cells were incubated with CD4-fluorescein isothiocyanate (FITC) antibody and CD25-PE antibody at 4 °C for 30 min in the dark. After fixation and permeabilization using reagents from Biolegend (San Diego, CA, USA), cells were stained with forkhead Box P3 (Foxp3)-Alexa Fluor 647 antibody (Biolegend) at 4 °C for 30 min in the dark. Cell analysis was performed using a flow cytometer (Beckman Coulter CytoFLEX, CA, USA), and data were analysed with FlowJo (Tree Star, Ashland, OR, USA).
Real-time fluorescence quantitative PCR
Total RNA was extracted from the ileum and colon using TRIzol Reagent (Invitrogen Life Technologies, Waltham, USA) according to the manufacturer’s instructions. Subsequently, cDNA was synthesized from the total RNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Wilmington, USA). The qPCR primers were synthesized by Invitrogen Biotechnology (Carlsbad, CA, USA). GAPDH served as the internal reference gene. PCR was carried out on a real-time quantitative PCR system (Bio-Rad, California, USA) with an initial step at 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 55 °C for 30 s. Subsequently, a melting curve was generated (65 °C for 5 s and 95 °C for 20 s). Relative gene expression levels were determined using the 2−ΔΔCT method. Primer sequences were as follows: GAPDH: 5’-GGG TGT GAA CCA CGA GAA AT-3’ and 5’-CCT TCC ACA ATG CCA AAG TT-3’; Foxp3: 5’-CAC CTT TCC AGA GTT CTT CCA CA-3’ and 5’-CGG ATG AGG GTG GCA TAG GT-3’; IL-17: 5’-GGA CTC TCC ACC GCA ATG AA-3’ and 5’-TTT CCC TCC GCA TTG ACA CA-3’. Each PCR was performed in technical triplicates.
DNA extraction and 16S rRNA gene sequencing
Faecal samples from each group were collected aseptically and stored at − 80 °C for future analysis. Faecal DNA was extracted using the E.Z.N.A.® soil DNA Kit (Omega Biotek, Norcross, GA, USA) according to the manufacturer’s instructions. The extracted DNA was assessed for quality on a 1% agarose gel, and DNA concentration and purity were determined using a NanoDrop 2000 UV‒vis spectrophotometer (Thermo Scientific). The V3-V4 region of the bacterial 16S rRNA gene was amplified using an ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA) with the primer pair 338F (5’-ACT CCT ACG GGA GGC AGC AG-3’) and 806R (5’-GGA CTA CHV GGG TWT CTA AT-3’). PCR conditions included an initial denaturation at 95 °C for 3 min, followed by 27 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s. The reaction concluded with a final extension step at 72 °C for 10 min. PCR amplicons were separated via 2% agarose gel electrophoresis and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA). Quantification of amplicons was performed using QuantiFluor-ST (Madison, Promega, WI, USA). Subsequently, the amplicons were pooled in equimolar concentrations for paired-end sequencing on an Illumina MiSeq PE300 platform by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China) according to the manufacturer’s instructions.
Microbiota data analysis
After demultiplexing, sequences underwent quality filtering with fastp (0.19.6)32 followed by merging using FLASH (v1.2.11). High-quality sequences were denoised with DADA233 in the QIIME234 (version 2020.2) pipeline with recommended parameters, leading to the generation of amplicon sequence variants (ASVs). To account for sequencing depth variability, rarefaction was applied, with all samples standardized to 20,000 sequences while maintaining an average Good’s coverage of 97.90%. Taxonomy assignment was performed by the naive Bayes consensus taxonomy classifier within QIIME2 and the SILVA 16S rRNA database (version 138). Bioinformatic analysis was conducted using the Majorbio Cloud platform (https://cloud.majorbio.com). Alpha diversity indices, including observed ASVs and the Shannon index, were computed using Mothur (version 1.30.1)35. Microbial community similarities were assessed using principal coordinate analysis (PCoA) based on Bray‒Curtis dissimilarity via the Vegan v2.5-3 package. The PERMANOVA test, also in Vegan v2.5-3, was used to determine treatment-related variation and significance. To identify significantly abundant taxa (phylum to genera) among groups (LDA score > 3, P < 0.05), we performed linear discriminant analysis effect size (LEfSe)36 (http://huttenhower.sph.harvard.edu/LEfSe).
Ethical approval and consent to participate
The study was approved by the Ethics Committee of Sichuan University West China Medical for Animal Experimentation (K2019002-2).